使用EPW计算孤立ME方程
时间:2023-11-03 10:37:02
1.自洽声子计算
在phonon目录下
scf.in
&control calculation = 'scf' restart_mode = 'from_scratch', prefix = 'pb', pseudo_dir = '../', outdir = './' / &system ibrav = 2, celldm(1) = 9.27, nat = 1, ntyp = 1, ecutwfc = 30.0 occupations = 'smearing', smearing = 'mp', degauss = 0.025 / &electrons diagonalization = 'david' mixing_beta = 0.7 conv_thr = 1.0d-12 / ATOMIC_SPECIES Pb 207.2 pb_s.UPF ATOMIC_POSITIONS Pb 0.00 0.00 0.00 K_POINTS {
automatic} 12 12 12 0 0 0 ph.in
-- &inputph prefix = 'pb', fildyn = 'pb.dyn.xml', fildvscf = 'dvscf' tr2_ph = 1.0d-17 ldisp = .true., nq1 = 3, nq2 = 3, nq3 = 3 / 2.转存有用的生成文件
在phonon目录下
python3 /work2/06868/giustino/EPW-SCHOOL/q-e/EPW/bin/pp.py 3.超导计算
nscf.in
&control calculation = 'nscf', restart_mode = 'from_scratc',
prefix = 'pb',
pseudo_dir = '../',
outdir = './',
verbosity = 'high'
/
&system
ibrav = 2,
celldm(1) = 9.27,
nat = 1,
ntyp = 1,
ecutwfc = 30.0,
occupations = 'smearing',
smearing = 'mp',
degauss = 0.025,
nbnd = 10,
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.0d-12
/
ATOMIC_SPECIES
Pb 207.2 pb_s.UPF
ATOMIC_POSITIONS crystal
Pb 0.000000000 0.000000000 0.000000000
K_POINTS crystal
27
0.00000000 0.00000000 0.00000000 3.703704e-02
0.00000000 0.00000000 0.33333333 3.703704e-02
0.00000000 0.00000000 0.66666667 3.703704e-02
0.00000000 0.33333333 0.00000000 3.703704e-02
0.00000000 0.33333333 0.33333333 3.703704e-02
0.00000000 0.33333333 0.66666667 3.703704e-02
0.00000000 0.66666667 0.00000000 3.703704e-02
0.00000000 0.66666667 0.33333333 3.703704e-02
0.00000000 0.66666667 0.66666667 3.703704e-02
0.33333333 0.00000000 0.00000000 3.703704e-02
0.33333333 0.00000000 0.33333333 3.703704e-02
0.33333333 0.00000000 0.66666667 3.703704e-02
0.33333333 0.33333333 0.00000000 3.703704e-02
0.33333333 0.33333333 0.33333333 3.703704e-02
0.33333333 0.33333333 0.66666667 3.703704e-02
0.33333333 0.66666667 0.00000000 3.703704e-02
0.33333333 0.66666667 0.33333333 3.703704e-02
0.33333333 0.66666667 0.66666667 3.703704e-02
0.66666667 0.00000000 0.00000000 3.703704e-02
0.66666667 0.00000000 0.33333333 3.703704e-02
0.66666667 0.00000000 0.66666667 3.703704e-02
0.66666667 0.33333333 0.00000000 3.703704e-02
0.66666667 0.33333333 0.33333333 3.703704e-02
0.66666667 0.33333333 0.66666667 3.703704e-02
0.66666667 0.66666667 0.00000000 3.703704e-02
0.66666667 0.66666667 0.33333333 3.703704e-02
0.66666667 0.66666667 0.66666667 3.703704e-02
nscf.in的最后的k坐标生成
/work2/06868/giustino/EPW-SCHOOL/q-e/wannier90-3.1.0/utility/kmesh.pl 3 3 3
没有这个文件夹证明你没安装EPW。请在QE文件夹中make epw。
epw1.in
--
&inputepw
prefix = 'pb',
amass(1) = 207.2
outdir = './'
dvscf_dir = '../phonon/save'
ep_coupling = .true.
elph = .true.
epwwrite = .true.
epwread = .false.
wannierize = .true.
nbndsub = 4
bands_skipped = 'exclude_bands = 1-5'
num_iter = 300
dis_froz_min= -3
dis_froz_max= 13.5
proj(1) = 'Pb:sp3'
wdata(1) = 'bands_plot = .true.'
wdata(2) = 'begin kpoint_path'
wdata(3) = 'G 0.00 0.00 0.00 X 0.00 0.50 0.50'
wdata(4) = 'X 0.00 0.50 0.50 W 0.25 0.50 0.75'
wdata(5) = 'W 0.25 0.50 0.75 L 0.50 0.50 0.50'
wdata(6) = 'L 0.50 0.50 0.50 K 0.375 0.375 0.75'
wdata(7) = 'K 0.375 0.375 0.75 G 0.00 0.00 0.00'
wdata(8) = 'G 0.00 0.00 0.00 L 0.50 0.50 0.50'
wdata(9) = 'end kpoint_path'
wdata(10) = 'bands_plot_format = gnuplot'
fsthick = 0.4 ! eV
degaussw = 0.1 ! eV
degaussq = 0.05 ! eV
ephwrite = .true.
eliashberg = .true.
liso = .true.
limag = .true.
lpade = .true.
lacon = .true.
nsiter = 500
npade = 40
conv_thr_iaxis = 1.0d-3
conv_thr_racon = 1.0d-3
wscut = 0.1 ! eV
muc = 0.1
temps = 0.3 0.9 1.5 2.1 2.7 3.3 3.9 4.2 4.4 4.5 4.6 4.65
nk1 = 3
nk2 = 3
nk3 = 3
nq1 = 3
nq2 = 3
nq3 = 3
mp_mesh_k = .true.
nkf1 = 18
nkf2 = 18
nkf3 = 18
nqf1 = 18
nqf2 = 18
nqf3 = 18
/
3.3生成的文件:
文件夹pb.ephmat
(1) ephmatXX 文件(每个 CPU 一个),其中包含密集 k 和 q 网格上费米窗 (fsthick) 内的电子-声子矩阵元素
(2) 包含密集 q 网格上声子频率的 freq 文件
(3) egnv 文件包含在密集 k 网格上的 Fermi 窗口内的特征值
(4) ikmap 文件,包含费米窗内密集(不可约)网格上的 k 点索引。
所有这些文件都是通过设置 ephwrite = .true. 生成的。这些文件是未格式化的,是求解 Migdal-Eliashberg 方程所必需的。
3.4计算过程
求解虚频率轴上的各向同性 Migdal-Eliashberg 方程。
这是通过设置关键字 eliashberg = .true.、liso = .true. 和 limag =.true 来实现的。 在输入文件中。 对于输入文件中指定的每个温度值【epw1.in中的temps】,方程自洽求解。 当达到收敛阈值 (conv thr iaxis) 或最大迭代次数 (nsiter) 时,每个温度下的计算结束。
注 1:如果在特定温度下达到最大迭代次数而没有达到收敛,代码将停止并且不会移动到列表中的下一个温度。
注 2:由于电子-声子矩阵元素不依赖于求解 Migdal Eliashberg 方程时的温度,因此可以在不同温度下的后续 EPW 计算中重复使用它们。这就是 pb.ephmat 目录保存ephmatXX的原因。
muc提供半经验库伦参数mu_star
在计算运行时,请注意 epw1.out 文件中的完整 EPW 运行所经历的不同步骤。 完成精细网格的插值后,代码将写入和读取求解 Migdal-Eliashberg 方程所需的文件,然后继续在指定温度下求解方程。
执行从虚频率轴到实频率轴的解的解析延展。 分析延续可以使用 Pad´e 近似值 (lpade =.true.) 或迭代过程 (lacon = .true.) 来完成。 迭代过程自洽地执行,直到达到收敛阈值 (conv thr racon) 或最大迭代次数 (nsiter)。
注意:如果在特定温度下达到最大迭代次数而没有达到收敛,代码将停止并且不会移动到列表中的下一个温度。
在计算结束时,您应该在每个给定温度下获得一些输出文件。
请注意,松原频率点的数量随着温度的升高而减少,因为频率 iωj = i(2n + 1)πT (n 整数) 小于截止频率 wscut。
3.5输出文件中的物理量
超导特性的计算将伴随着大量的 I/O。 下面我们将描述保存在输出文件中的各种物理量以及如何处理它们。 我们将在输出文件的名称中使用 XX 来表示求解方程的温度。
a.Eliashberg 谱函数和积分电子-声子耦合强度 (λ)
存在文件pb.a2f, pb.a2f iso, and pb.a2f proj中。
需要设置eliashberg = .true.
pb.a2f中包含 the isotropic Eliashberg spectral function α2F(ω)和积分电子 - 声子耦合强度(cumulative electron-phonon coupling strength) λ 作为频率 ω (meV) 的函数对于每一个phonon smearing。(关于smearing的信息在文档末尾有介绍。)
472 / 5,000
翻译结果
pb.a2f iso 和 pb.a2f proj 文件包含作为频率 ω (meV) 函数的 Eliashberg 谱函数,其中两个文件中的第 2 列是对应于 pb.a2f 中第一个拖尾的 Eliashberg 谱函数。pb.a2f proj 中剩余的(3×原子数)列包含与 pb.a2f 中的第一个拖尾相对应的模式分辨的 Eliashberg 光谱函数(没有关于哪些模式对应于哪些原子种类的具体信息)。
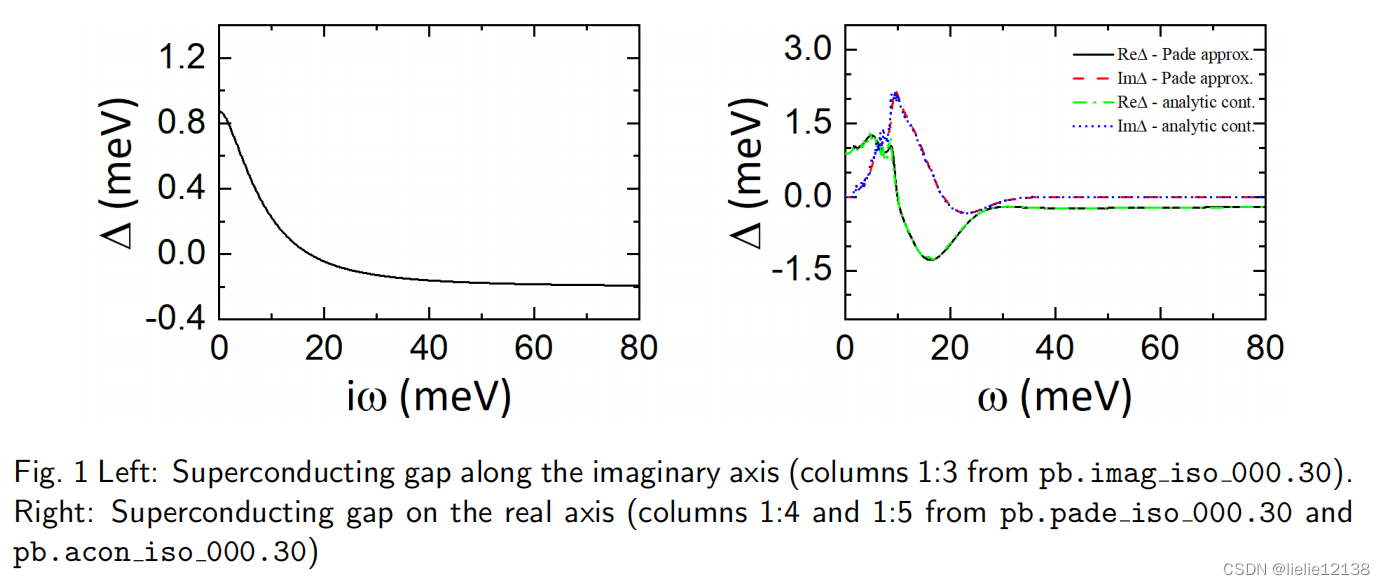
b.沿虚频轴和实频轴的超导gap。
pb.imag iso XX 文件是通过设置 eliashberg = .true.、liso = .true. 和 limag = .true. 生成的。每个文件包含 4 列:沿虚轴的松原频率 iωj (eV)、准粒子重整化 函数 Z(iωj )、超导间隙 Δ(iωj ) (eV) 和正常状态下的准粒子重整化函数 ZN (iωj )。
pb.pade iso XX 文件是通过设置 lpade = .true… 生成的。每个文件包含 5 列:沿实轴的频率 ω (eV)、准粒子重整化函数 ReZ(ω) 的实部、虚部 准粒子重整化函数 ImZ(ω)、超导间隙 ReΔ(ω) (eV) 的实部和超导间隙 ImΔ(ω) (eV) 的虚部。
pb.acon iso XX 文件是通过设置 lacon = .true 生成的。 并包含与 pb.pade iso XX 类似的信息。
4.沿虚频率轴和实频率轴绘制超导间隙
您可以使用以下 gnuplot 脚本来绘制 pb.imag iso 000.30、pb.pade iso 000.30 和 pb.acon iso 000.30。 您应该在 0.3 K 处获得类似于图 1 的内容。
注意单位*1000
$ gnuplot
gnuplot> set xlabel "iw (meV)"
gnuplot> set ylabel "Delta (meV)"
gnuplot> plot "pb.imag_iso_000.30" u ($1*1000):($3*1000) w l lw 2 lt rgb "black" \
> title "Delta-Imag"
gnuplot> set xlabel "w (meV)"
gnuplot> set ylabel "Delta (meV)"
gnuplot> plot "pb.pade_iso_000.30" u ($1*1000):($4*1000) w l lw 1 lt rgb "black" \
> title "Re(Delta)-Pade", \
> "" u ($1*1000):($5*1000) w l lw 1 lt rgb "red" title "Im(Delta)-Pade", \
> "pb.acon_iso_000.30" u ($1*1000):($4*1000) w l lw 1 lt rgb "blue" \
> title "Re(Delta)-analytic cont.", \
> "" u ($1*1000):($5*1000) w l lw 1 lt rgb "green" \
> title "Im(Delta)-analytic cont."
5.绘制超导间隙的前沿作为温度的函数。
使用 shell 脚本 gap0 imag(也如下所示)提取超导间隙的前沿作为温度的函数,并将信息保存在新文件 pb.imag iso gap0 中。
建立脚本gap0_imag.sh
#!/bin/tcsh
awk 'FNR==2 {print FILENAME,$0}' pb.imag_iso_* | awk '{print $1 " " $4*1000}' > pb.imag_iso_gap0
sed -i 's/pb.imag_iso_//' pb.imag_iso_gap0
运行脚本
Bash gap0_imag.sh
您可以使用以下 gnuplot 脚本来绘制 pb.imag iso gap0文件。 你应该得到类似于图 3 的东西
$ gnuplot
gnuplot> set xlabel "T (K)"
gnuplot> set ylabel "Delta_0 (meV)"
gnuplot> set xrange [0:6]
gnuplot> set yrange [0:1.2]
gnuplot> plot "pb.imag_iso_gap0" with lp ls 3 notitle
您可以从实轴上的计算中进一步提取超导间隙的前沿作为温度的函数,并将其与图 3 所示的虚轴上获得的值进行比较。您可以使用 shell 脚本script gap0 pade 和 script gap0 acon 以获取 pb.pade iso gap0 和 pb.acon iso gap0 文件。 接下来使用 gnuplot 绘制这些文件,就像上面对 pb.imag iso gap0 所做的那样。
$ ./script_gap0_pade
#!/bin/tcsh
awk 'FNR==2 {print FILENAME,$0}' pb.pade_iso_* | awk '{print $1 " " $5*1000}' > pb.pade_iso_gap0
sed -i 's/pb.pade_iso_//' pb.pade_iso_gap0
./script_gap0_acon
#!/bin/tcsh
awk 'FNR==2 {print FILENAME,$0}' pb.acon_iso_* | awk '{print $1 " " $5*1000}' > pb.acon_iso_gap0
sed -i 's/pb.acon_iso_//' pb.acon_iso_gap0
6. 求解the linearized isotropic Eliashberg equations的临界温度
从包含 Eliashberg 谱函数 (pb.a2f iso) 的文件开始来完成此步骤:
作业脚本为:
ibrun $PATHQE/bin/epw.x -nk 1 -in epw2.in > epw2.out
输入文件为(仅显示与 epw1.in 文件的差异):
ep_coupling = .false.
elph = .false.
epwwrite = .false.
epwread = .true.
wannierize = .false.
ephwrite = .false.
fila2f = 'pb.a2f_iso'
lpade = .false.
lacon = .false.
tc_linear = .true. ! solve linearized ME eqn. for Tc
tc_linear_solver = 'power' ! algorithm to solve Tc eigenvalue problem: 'power' OR 'lapack'
nstemp = 21 ! number of temperature points
temps = 0.25 5.25 ! evenly spaced nstemp temperature points according to
! (temps(2)-temps(1))/(nstemp-1).
注意1:在这种情况下,不使用 ephmatXX、freq、egnv 和 ikmap 文件(保存在 prefix.ephmat 目录中)。 您还可以从包含 Eliashberg 谱函数 (pb.a2f iso) 的文件开始求解其他温度下的各向同性 Migdal-Eliashberg 方程。 此过程不适用于求解各向异性 Migdal-Eliashberg 方程。
注意2:如果从 Eliashberg 谱函数开始求解各向同性 Migdal Eliashberg 或线性化 Eliashberg 方程,则只需使用一个 CPU
您可以使用脚本 max eigenvalue 从 epw2.out 中提取作为温度函数的最大特征值,并将数据保存在 data max eigenvalue.dat 文件中。
$ ./script_max_eigenvalue
#!/bin/tcsh
grep -A 25 "Max. eigenvalue" epw2.out | tail -21 | awk '{print $1 " " $2}' > data_max_eigenvalue.dat
绘制数据 max eigenvalue.dat 以获得 Tc。 临界温度定义为最大特征值接近 1 的值。您可以使用下面的 gnuplot 脚本得到图 4 所示的图形
$ gnuplot
gnuplot> set xlabel "T (K)"
gnuplot> set ylabel "Max. eigenvalue"
gnuplot> set xrange [0:6]
gnuplot> set arrow from 0,1 to 6,1 nohead lt 2 lw 1
gnuplot> plot "data_max_eigenvalue.dat" with lp ls 3 notitle
7.epw.in中部分参数的选择
https://www.wanniertools.org/tutorials/high-quality-wfs/
在我们从第一性原理计算中获得自洽电荷密度后,高质量的 Wannier 函数 (WFs) 对物理性质计算非常有用。 但是,对于初学者来说,要获得好的WF并不容易。 在这里,我们想介绍一种标准的方法,用第一性原理软件包和软件Wannier90构建高质量的Wannier函数。 在这里,我们不关注构建最大局部化 Wannier 函数 (MLWF)。 我们只想构建一个基于WFs的良好紧束缚模型,该模型能够在我们感兴趣的能量范围内再现能带结构。以下是高质量WFs的四个标准。
-
完美拟合您感兴趣的能量范围内的DFT能带(band)。
-
保持原子轨道对称
-
Well localized
-
As small number of WFs as possible尽量少的wannier波函数
这里有三个步骤可以帮助您实现这四个目标 -
Choose projectors
-
Choose disentanglement(解开?) energy window.
-
Choose frozen energy window.
我想以 Bi2Se3 为例。 为了节省计算成本,我们没有考虑自旋轨道耦合(SOC)效应。
1.Choose projectors
让我们先进行能带分析。 图 1 是未考虑 SOC 的 Bi2Se3 能带结构。 绿色阴影部分是我们关心的能量范围。我们想构建一个紧密结合的模型,可以很好地再现感兴趣范围内的能带。 但是,这些频段与下方和上方的其他频段相连。 值得注意的是,-8.4eV 和 -6eV 附近有很大的gap。 这是一个好兆头,因为该间隙下方的条带未与我们感兴趣的条带杂交。在这种情况下,间隙下方的那些条带不必在我们的紧密结合模型中呈现。 最终,WF 的数量会减少。 它满足第四个标准。
在初步了解能带结构后,我们仍然需要弄清楚感兴趣的能量范围内能带的特征是什么,并找出哪些能带与它们高度杂化。这一步称为“fatband analysis(胖带分析)”,我们需要将原子轨道投影到布洛赫波函数上,这称为投影权重(the projected weight)。
我们将 Bloch 波函数投影到 Bi 和 Se 的 s、p、d 原子轨道上(这一步是要怎么做呢)。 结果如图2所示。图2(a)和(d)表明[-15,-8]eV之间的能带由铋和硒的s轨道支配。图2(c)和(f)显示Bi和Se的d轨道位于5eV以上。 图 2 (b) 和 (e) 表明能量范围 [-6, 5] eV 由 Bi 和 Se 的 p 轨道支配。我们感兴趣的能量范围,在图 1 中以绿色阴影表示,就在这个能量范围内。 尽管 p 和 d 轨道之间在 4eV 左右存在能带重叠,但我们仍然可以忽略 d 轨道的贡献,因为它们与 p 轨道不高度杂化,并且可以通过能带结构的一些变形而移开。
在“fatband analysis”之后,我们选择 Bi 和 Se 的 p 轨道作为 WFs 的projectors。 那么将有(2个Bi原子)(3个p轨道)+(3个Se原子)(3个p轨道)=15个原子轨道,总共15个没有SOC的WF。
begin projections
Bi : p
Se : p
end projections
2.Choose disentanglement energy window dis_win
isentanglement energy window 是提取 the target bands的 window。这里有两个规则去选择这个窗口。
- 不只the target bands应该包含在这个窗口中, the major weight of the projectors 也应该包含在这个窗口中。
- 该窗口应尽可能小,以降低计算成本。
图三的彩色阴影给出了窗口的三种选择。(这个图是怎么画出来的呀。。。)图 3(a) 是正确的选择。 图 3(b) 是一个非常糟糕的选择,因为虚线圆圈标记处可以看出缺少一个带,这个带的weight comes from the projectors。如果缺少此权重,则 WF 的分布可能会变大(See Fig 4b and 4e)。图 3© 显示了一个不好的选择,因为它包含不包含投影特征的不必要的波段。
Choose frozen energy window froz_win
The frozen window is a window used for disentanglement procedure.Bloch 态不会随其他带旋转出此窗口。选择froz_win 有三个规则。 1) 在 froz_win 中,除了投影仪之外,不应有来自其他轨道的权重(贡献)。 2) 纯粹来自投影仪的能量范围应包含在 froz_win 中。 3)froz_win 应该尽可能的大。
图 4 显示了三种选择。 (a) 是一个不错的选择,因为它满足三个规则。 (b) 是错误的选择,因为它违反了第一条规则。 © 是正确的选择,但不是完美的选择,因为它不遵循规则 2 和 3。
3.结论
经过上述过程,您将获得非常好的WF,可能性很大。 图 5 显示了 DFT(黑线)和 Wannier 插值(红点)之间的能带结构比较。 它显示了完美的一致性。
(所以wannier插值的目的就是为了插值出能带结构???)
最后,我们想分享一些其他的技巧。
- dis_num_iter 最好不要设置的太大,大部分时候200就够了。 如果它太大,那么布洛赫频带可能会过度混合。
- 如果你想要类似原子的 WF,最好将 num_iter 设置为小于 20。大量的 num_iter 会混合 WF,使得输出的 WF 不是原子的。
- 如果您发现 WF 的分布非常大或在 Wannier 插值频带中有一些摆动,请尝试仔细调整投影仪、dis_win 和 froz_win。
(在epw.in中这两个量是不分开的是dis_froz_win,确认一下这个是什么)
VASP 用户专用:
如果您要为带有 SOC 的系统构建 WF,如果存在“LWANNIER90 = T”,请在 INCAR 文件中设置“ISYM=-1”。 由于在 VASP 中将旋量波函数从一个 k 点复制到与对称相关的一个 k 点存在一些错误,因此我们关闭此对称标签后,WFs 的质量将显着提高。


